

Basic principles of genetics
What is a genetic disease?
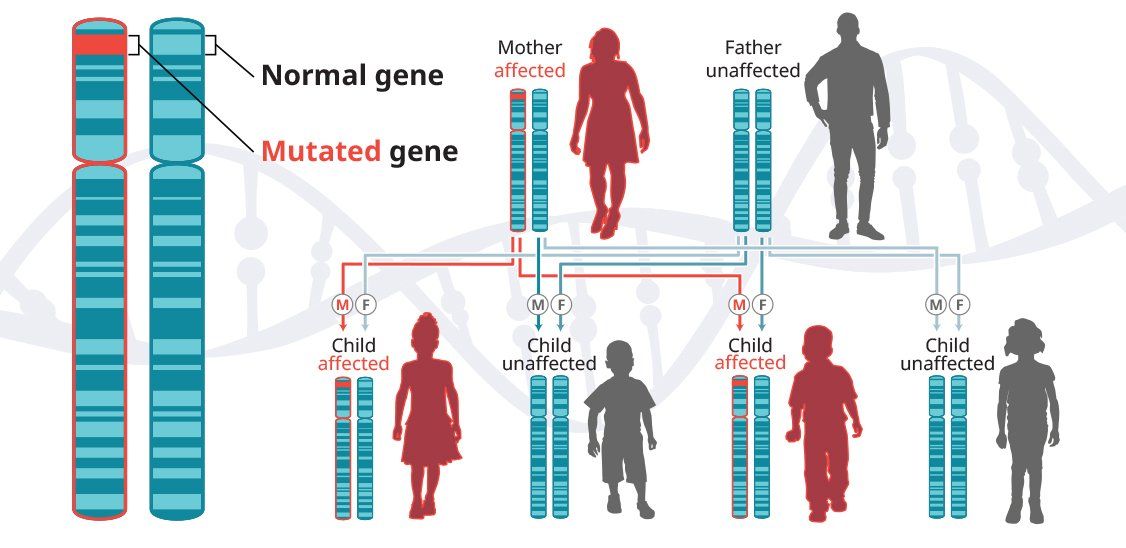
In autosomal-dominant diseases, one copy of the defective gene is sufficient for a person to inherit the condition. In some cases, an affected child inherits the disease from an affected parent.5
Examples of autosomal-dominant diseases
Huntington’s disease (HD)
Huntington’s disease is a progressive neurodegenerative disease that is inherited in an autosomal-dominant pattern, which means that a person inherits one defective copy of the gene and can develop the disease.6-8 When a man or a woman with HD has children, each child has a 50% chance of inheriting the mutated gene and developing the disease. In the United States, approximately 30,000 people have HD and another 200,000 are at risk of developing the disease.9
Spinocerebellar ataxias (SCAs)
Spinocerebellar ataxias (SCAs) are a group of inherited progressive neurodegenerative diseases that mainly affect the cerebellum. Currently, more than 40 types of SCAs have been identified. Not all SCAs are restricted to pure cerebellar degeneration; some types involve other areas of the central nervous system, including the pontine nuclei, spinal cord, cortex, peripheral nerves, and basal ganglia.15
The prevalence of SCAs, which are inherited in an autosomal-dominant fashion, is estimated to be about 1 to 5 in 100,000 individuals.16
Spinocerebellar ataxia 3 (SCA3) is the most common subtype of type 1 autosomal-dominant ataxias worldwide. The prevalence of SCA3 is estimated to be 1 to 2 in 100,000 people but varies significantly based on geography and ethnicity.17
X-linked recessive diseases
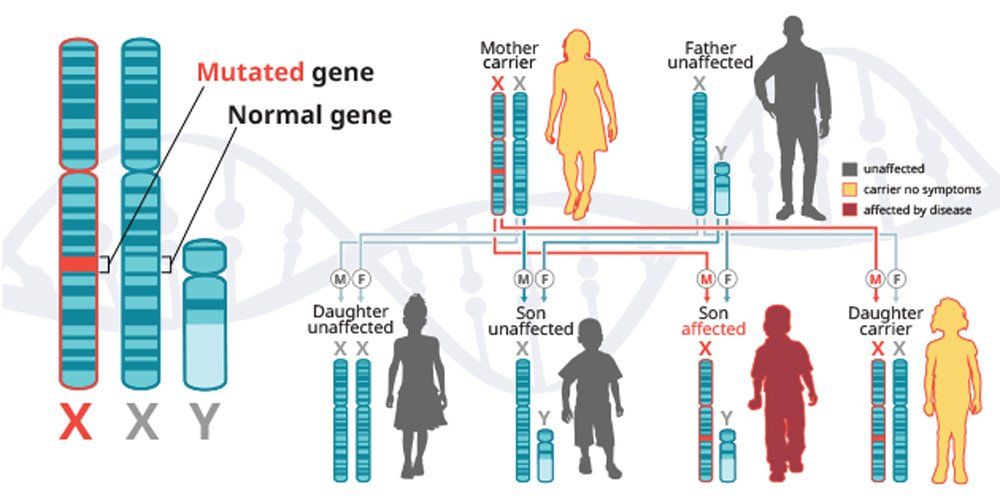
X-linked recessive diseases are caused by mutations in genes on the X chromosome. Men only have one X chromosome, so only one defective copy of the gene is sufficient to cause disease. Women have two X chromosomes, so a mutation would have to occur in both copies of the gene to cause disease; although, women carriers can sometimes display some disease symptoms. It is less likely that women will have two defective copies of this gene. Men are affected by X-linked recessive diseases much more frequently than women. In fact, one characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.5
Examples of X-linked recessive diseases
Fabry disease
Fabry disease is caused by mutations in the
GLA gene, which is located on the X chromosome, one of two sex chromosomes. Thus, Fabry is an
X-linked disease, inherited in a recessive pattern.
Fabry disease occurs in all populations and affects both males and females. It is estimated that there are about 3800 men with Fabry disease in the United States. It is not known how many women are affected by the disease.22
Published data from the Fabry registry indicates that men with the disease are expected to live to an average age of 50 to 57 years. Women with Fabry disease are expected to live to an average age of 64 to 72 years.24
Hemophilia
Hemophilia is a bleeding disorder that interferes with
blood clotting. Hemophilia A and hemophilia B are inherited in an
X-linked recessive pattern. This means that the genes associated with hemophilia are located on the X chromosome, one of two sex chromosomes.25
For all severities of hemophilia A, it is estimated that this disease occurs in 17.1 cases per 100,000 men. Hemophilia B is not as common. For all severities of hemophilia B, it is estimated that this disease occurs in 3.8 cases per 100,000 men.26